'Rogue' Chinese Virologist Joins Twitter, Publishes "Smoking Gun" Evidence COVID-19 Created In Lab
On Saturday we reported that Dr. Li-Meng Yan - a Chinese virologist (MD, PhD) who fled the country, leaving her job at a prestigious Hong Kong university - appeared last week on British television where she claimed SARS-CoV-2, the virus which causes COVID-19, was created by Chinese scientists in a lab.

On Sunday, Li-Meng joined Twitter - and on Monday, just hours ago, she tweeted a link to a paper she co-authored with three other Chinese scientists titled:
Unusual Features of the SARS-CoV-2 Genome Suggesting Sophisticated Laboratory Modification Rather Than Natural Evolution and Delineation of Its Probable Synthetic Route
— Dr. Li-Meng YAN (@LiMengYAN119) September 14, 2020
https://archive.vn/wip/KxJJr
She also posted a link to her credentials on ResearchGate, revealing
her (prior?) affiliation with The University of Hong Kong and 13
publications which have been cited 557 times.
— Dr. Li-Meng YAN (@LiMengYAN119) September 14, 2020https://archive.vn/wip/GD3hV
Cutting to the chase:
"The evidence shows that SARS-CoV-2 should be a laboratory product created by using bat coronaviruses ZC45 and/or ZXC21 as a template and/or backbone. Building upon the evidence, we further postulate a synthetic route for SARS-CoV-2, demonstrating that the laboratory-creation of this coronavirus is convenient and can be accomplished in approximately six months.
Here is the extended punchline:
The receptor-binding motif of SARS-CoV-2 Spike cannot be born from nature and should have been created through genetic engineering.
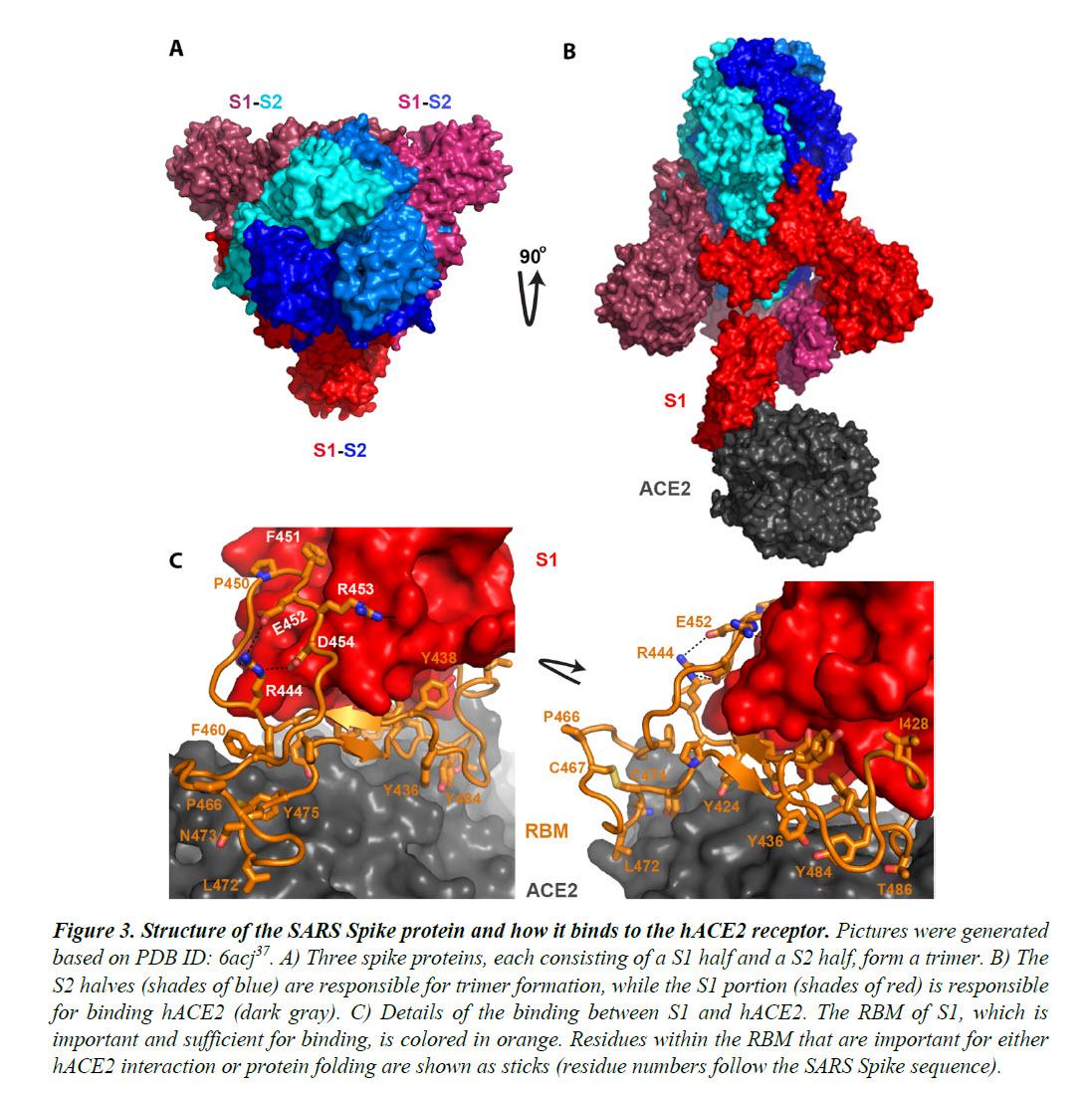
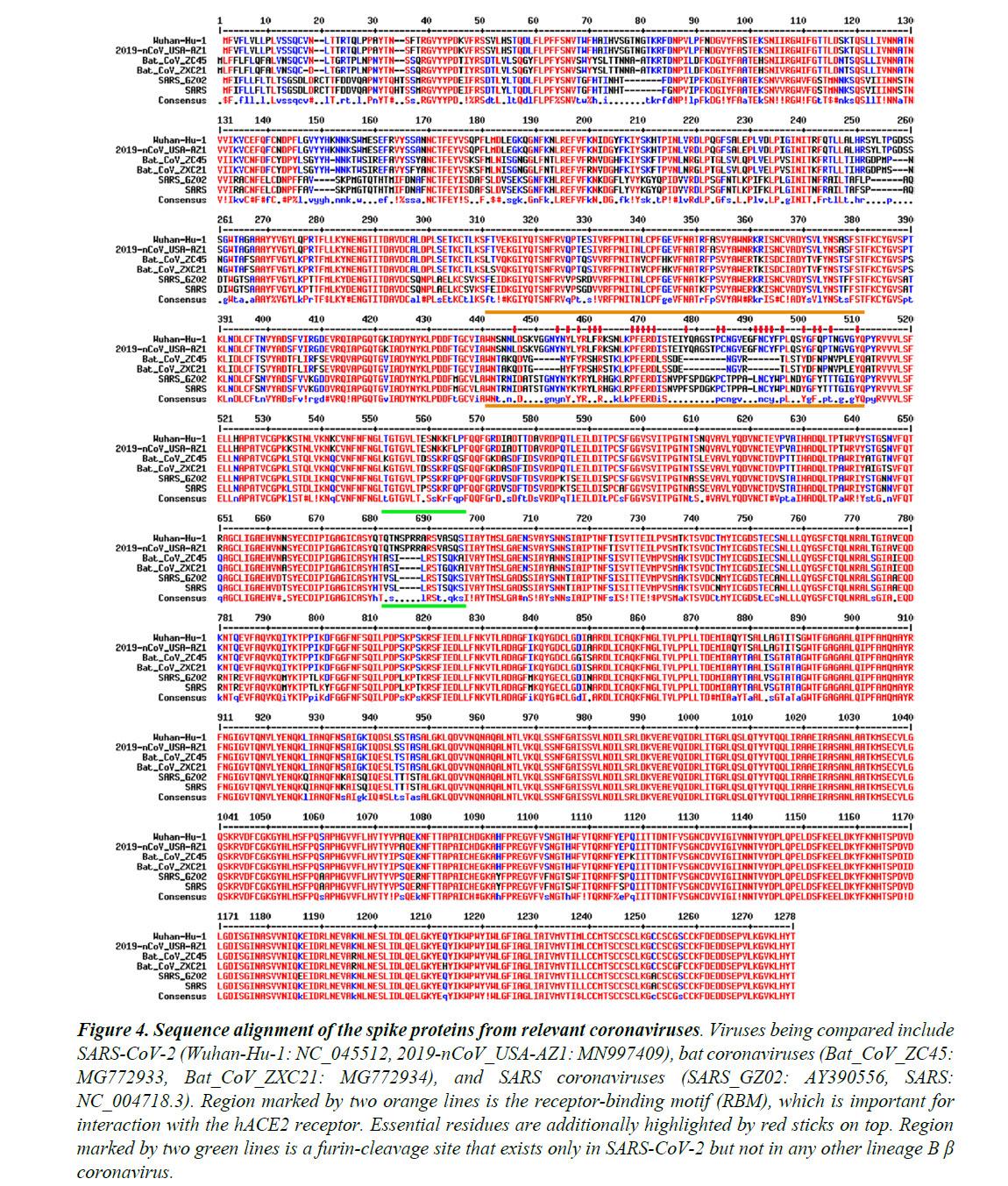
The Spike proteins decorate the exterior of the coronavirus particles. They play an important role in infection as they mediate the interaction with host cell receptors and thereby help determine the host range and tissue tropism of the virus. The Spike protein is split into two halves (Figure 3). The front or N-terminal half is named S1, which is fully responsible for binding the host receptor. In both SARS-CoV and SARS-CoV-2 infections, the host cell receptor is hACE2. Within S1, a segment of around 70 amino acids makes direct contacts with hACE2 and is correspondingly named the receptor-binding motif (RBM) (Figure 3C). In SARS-CoV and SARS-CoV-2, the RBM fully determines the interaction with hACE2. The C-terminal half of the Spike protein is named S2. The main function of S2 includes maintaining trimer formation and, upon successive protease cleavages at the S1/S2 junction and a downstream S2’ position, mediating membrane fusion to enable cellular entry of the virus.
Similar to what is observed for other viral proteins, S2 of SARS-CoV-2 shares a high sequence identity (95%) with S2 of ZC45/ZXC21. In stark contrast, between SARS-CoV-2 and ZC45/ZXC21, the S1 protein, which dictates which host (human or bat) the virus can infect, is much less conserved with the amino acid sequence identity being only 69%.
Figure 4 shows the sequence alignment of the Spike proteins from six β coronaviruses. Two are viruses isolated from the current pandemic (Wuhan-Hu-1, 2019-nCoV_USA-AZ1); two are the suspected template viruses (Bat_CoV_ZC45, Bat_CoV_ZXC21); two are SARS coronaviruses (SARS_GZ02, SARS). The RBM is highlighted in between two orange lines. Clearly, despite the high sequence identity for the overall genomes, the RBM of SARS-CoV-2 differs significantly from those of ZC45 and ZXC21. Intriguingly, the RBM of SARS-CoV-2 resembles, on a great deal, the RBM of SARS Spike. Although this is not an exact “copy and paste”, careful examination of the Spike-hACE2 structures37,38 reveals that all residues essential for either hACE2 binding or protein folding (orange sticks in Figure 3C and what is highlighted by red short lines in Figure 4) are “kept”.
Most of these essential residues are precisely preserved, including those involved in disulfide bond formation (C467, C474) and electrostatic interactions (R444, E452, R453, D454), which are pivotal for the structural integrity of the RBM (Figure 3C and 4). The few changes within the group of essential residues are almost exclusively hydrophobic “substitutions” (I428àL, L443àF, F460àY, L472àF, Y484àQ), which should not affect either protein folding or the hACE2-interaction. At the same time, majority of the amino acid residues that are non-essential have “mutated” (Figure 4, RBM residues not labeled with short red lines). Judging from this sequence analysis alone, we were convinced early on that not only would the SARS-CoV-2 Spike protein bind hACE2 but also the binding would resemble, precisely, that between the original SARS Spike protein and hACE223. Recent structural work has confirmed our prediction.
As elaborated below, the way that SARS-CoV-2 RBM resembles SARS-CoV RBM and the overall sequence conservation pattern between SARS-CoV-2 and ZC45/ZXC21 are highly unusual. Collectively, this suggests that portions of the SARS-CoV-2 genome have not been derived from natural quasi-species viral particle evolution.
The paper then makes two critical observations for those who claim that SARS-CoV-2 has a natural origin: its RBM could have only been acquired in one of the two possible routes:
1) an ancient recombination event followed by convergent evolution or
2) a natural recombination event that occurred fairly recently.
She first dismisses option 1:
"this convergent evolution process would also result in the accumulation of a large amount of mutations in other parts of the genome, rendering the overall sequence identity relatively low. The high sequence identity between SARS-CoV-2 and ZC45/ZXC21 on various proteins (94-100% identity) do not support this scenario and, therefore, clearly indicates that SARS-CoV- 2 carrying such an RBM cannot come from a ZC45/ZXC21-like bat coronavirus through this convergent evolutionary route."
She then dismisses option 2:
In the second scenario, the ZC45/ZXC21-like coronavirus would have to have recently recombined and swapped its RBM with another coronavirus that had successfully adapted to bind an animal ACE2 highly homologous to hACE2. The likelihood of such an event depends, in part, on the general requirements of natural recombination: 1) that the two different viruses share significant sequence similarity; 2) that they must co-infect and be present in the same cell of the same animal; 3) that the recombinant virus would not be cleared by the host or make the host extinct; 4) that the recombinant virus eventually would have to become stable and transmissible within the host species.
In regard to this recent recombination scenario, the animal reservoir could not be bats because the ACE2 proteins in bats are not homologous enough to hACE2 and therefore the adaption would not be able to yield an RBM sequence as seen in SARS-CoV-2. This animal reservoir also could not be humans as the ZC45/ZXC21-like coronavirus would not be able to infect humans. In addition, there has been no evidence of any SARS-CoV-2 or SARS-CoV-2-like virus circulating in the human population prior to late 2019. Intriguingly, according to a recent bioinformatics study, SARS-CoV-2 was well-adapted for humans since the start of the outbreak.
Which leaves just one option:
Only one other possibility of natural evolution remains, which is that the ZC45/ZXC21-like virus and a coronavirus containing a SARS-like RBM could have recombined in an intermediate host where the ACE2 protein is homologous to hACE2. Several laboratories have reported that some of the Sunda pangolins smuggled into China from Malaysia carried coronaviruses, the receptor-binding domain (RBD) of which is almost identical to that of SARS-CoV-227-29,31. They then went on to suggest that pangolins are the likely intermediate host for SARS-CoV-227-29,31. However, recent independent reports have found significant flaws in this data40-42. Furthermore, contrary to these reports27-29,31, no coronaviruses have been detected in Sunda pangolin samples collected for over a decade in Malaysia and Sabah between 2009 and 201943. A recent study also showed that the RBD, which is shared between SARS-CoV-2 and the reported pangolin coronaviruses, binds to hACE2 ten times stronger than to the pangolin ACE22, further dismissing pangolins as the possible intermediate host. Finally, an in silico study, while echoing the notion that pangolins are not likely an intermediate host, also indicated that none of the animal ACE2 proteins examined in their study exhibited more favorable binding potential to the SARS-CoV-2 Spike protein than hACE2 did. This last study virtually exempted all animals from their suspected roles as an intermediate host, which is consistent with the observation that SARS-CoV-2 was well-adapted for humans from the start of the outbreak. This is significant because these findings collectively suggest that no intermediate host seems to exist for SARS-CoV-2, which at the very least diminishes the possibility of a recombinant event occurring in an intermediate host.
Fast-forwarding to the smoking gun:
Given that RBM fully dictates hACE2-binding and that the SARS RBM-hACE2 binding was fully characterized by high-resolution structures (Figure 3)37,38, this RBM-only swap would not be any riskier than the full Spike swap. In fact, the feasibility of this RBM-swap strategy has been proven. In 2008, Dr. Zhengli Shi’s group swapped a SARS RBM into the Spike proteins of several SARS-like bat coronaviruses after introducing a restriction site into a codon-optimized spike gene (Figure 5C). They then validated the binding of the resulted chimeric Spike proteins with hACE2. Furthermore, in a recent publication, the RBM of SARS-CoV-2 was swapped into the receptor-binding domain (RBD) of SARSCoV, resulting in a chimeric RBD fully functional in binding hACE2 (Figure 5C)39. Strikingly, in both cases, the manipulated RBM segments resemble almost exactly the RBM defined by the positions of the EcoRI and BstEII sites (Figure 5C). Although cloning details are lacking in both publications39,47, it is conceivable that the actual restriction sites may vary depending on the spike gene receiving the RBM insertion as well as the convenience in introducing unique restriction site(s) in regions of interest. It is noteworthy that the corresponding author of this recent publication, Dr. Fang Li, has been an active collaborator of Dr. Zhengli Shi since 201049-53. Dr. Li was the first person in the world to have structurally elucidated the binding between SARS-CoV RBD and hACE238 and has been the leading expert in the structural understanding of Spike-ACE2 interactions. The striking finding of EcoRI and BstEII restriction sites at either end of the SARS-CoV-2 RBM, respectively, and the fact that the same RBM region has been swapped both by Dr. Shi and by her long-term collaborator, respectively, using restriction enzyme digestion methods are unlikely a coincidence. Rather, it is the smoking gun proving that the RBM/Spike of SARS-CoV-2 is a product of genetic manipulation."
It gets better, because the Chinese scientists then presciently tried to cover their tracks:
Although it may be convenient to copy the exact sequence of SARS RBM, it would be too clear a sign of artificial design and manipulation. The more deceiving approach would be to change a few nonessential residues, while preserving the ones critical for binding. This design could be well-guided by the high-resolution structures (Figure 3)37,38. This way, when the overall sequence of the RBM would appear to be more distinct from that of the SARS RBM, the hACE2-binding ability would be well-preserved. We believe that all of the crucial residues (residues labeled with red sticks in Figure 4, which are the same residues shown in sticks in Figure 3C) should have been “kept”. As described earlier, while some should be direct preservation, some should have been switched to residues with similar properties, which would not disrupt hACE2-binding and may even strengthen the association further [ZH: i.e., the virus was weaponized and enchanced]. Importantly, changes might have been made intentionally at non-essential sites, making it less like a “copy and paste” of the SARS RBM.
Yan also discusses the infamous furin-cleavage site:
... a close examination of the nucleotide sequence of the furin-cleavage site in SARS-CoV-2 spike has revealed that the two consecutive Arg residues within the inserted sequence (- PRRA-) are both coded by the rare codon CGG (least used codon for Arg in SARS-CoV-2) (Figure 7).
In fact, this CGGCGG arrangement is the only instance found in the SARS-CoV-2 genome where this rare codon is used in tandem. This observation strongly suggests that this furin-cleavage site should be a result of genetic engineering. Adding to the suspicion, a FauI restriction site is formulated by the codon choices here, suggesting the possibility that the restriction fragment length polymorphism, a technique that a WIV lab is proficient at, could have been involved. There, the fragmentation pattern resulted from FauI digestion could be used to monitor the preservation of the furin-cleavage site in Spike as this furin-cleavage site is prone to deletions in vitro. Specifically, RT-PCR on the spike gene of the recovered viruses from cell cultures or laboratory animals could be carried out, the product of which would be subjected to FauI digestion. Viruses retaining or losing the furin-cleavage site would then yield distinct patterns, allowing convenient tracking of the virus(es) of interest.

And another critical allegation: once again, the Wuhan Researchers were doing everything in their power to weaponize and boost the "enhancement of the infectivity and pathogenicity of the laboratory-made coronavirus":
The evidence collectively suggests that the furin-cleavage site in the SARS-CoV-2 Spike protein may not have come from nature and could be the result of genetic manipulation. The purpose of this manipulation could have been to assess any potential enhancement of the infectivity and pathogenicity of the laboratory-made coronavirus.
Summarizing the above:
Evidence presented in this part reveals that certain aspects of the SARS-CoV-2 genome are extremely difficult to reconcile to being a result of natural evolution. The alternative theory we suggest is that the virus may have been created by using ZC45/ZXC21 bat coronavirus(es) as the backbone and/or template. The Spike protein, especially the RBM within it, should have been artificially manipulated, upon which the virus has acquired the ability to bind hACE2 and infect humans. This is supported by the finding of a unique restriction enzyme digestion site at either end of the RBM. An unusual furin-cleavage site may have been introduced and inserted at the S1/S2 junction of the Spike protein, which contributes to the increased virulence and pathogenicity of the virus.
These transformations have then staged the SARS CoV-2 virus to eventually become a highly-transmissible, onset-hidden, lethal, sequelae-unclear, and massively disruptive pathogen.
Evidently, the possibility that SARS-CoV-2 could have been created through gain-of-function manipulations at the WIV is significant and should be investigated thoroughly and independently.
Finally, those curious how the virus could have been created synthetically in Wuhan, here is a diagram proposed by Dr. Yan explaining all the required steps:
Her full paper is below:
And for those who missed it, here's Li-Meng's interview on UK television:
https://www.zerohedge.com/medical/rogue-chinese-virologist-joins-twitter-publishes-evidence-covid-19-created-lab
___
Wow Chinese Virologist Dr. Li-Meng Yan Blows Whistle on China “Manufacturing” COVID-19…
In a stunning segment on Tucker Carlson tonight, Chinese virologist Dr. Li-Meng Yan explains how China actually manufactured the COVID-19 virus by weaponizing and modifying the genetic sequence within the China bat virus.
According to Dr. Li-Meng Yan the virus was specifically created and released by China.